Pneumonia Intersticial Linfocítica
 Créditos:
Dra. Elazir Mota - Rio de Janeiro/RJ
Créditos:
Dra. Elazir Mota - Rio de Janeiro/RJ
Descrição das figuras:
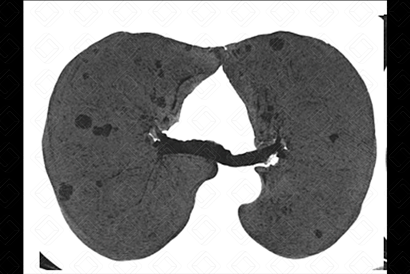
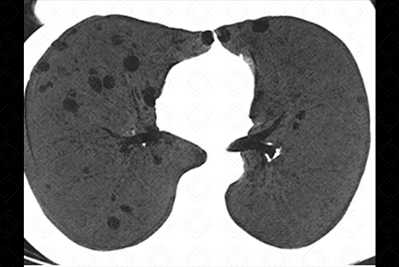
Tomografia computadorizada com projeção em intensidade mínima - MnIP (mulher, 64 anos, portadora de síndrome de Sjögren). Múltiplos cistos, menores que 10 mm, de paredes finas e de distribuição bilateral.
Pneumonia intersticial linfocítica (PIL):
É
uma doença pulmonar caracterizada por infiltração intersticial difusa, composta por uma combinação de linfócitos reativos T e B, e plasmócitos.
Frequentemente ocorre em associação com outras doenças de base, principalmente a
síndrome de
Sjögren
e a infecção pelo vírus da imunodeficiência humana
(HIV)
, sendo a forma idiopática da PIL bastante rara. Outras doenças reumatológicas, como a artrite reumatoide e o lúpus eritematoso sistêmico, também podem estar associadas. Dentre outras causas infecciosas, além da infecção pelo HIV, destacam-se as infecções pelos vírus Epstein-Barr (EBV) e vírus linfotrópico de células T humanas (HTLV).
A PIL relacionada ao HIV ocorre mais comumente na faixa pediátrica, sendo considerada uma doença definidora de síndrome de imunodeficiência adquirida (SIDA) em indivíduos com idade inferior a 13 anos. Cerca de 16 a 50% das crianças infectadas com HIV cursam com desenvolvimento de PIL no segundo ou terceiro ano de vida.
Quadro clínico: O curso clínico da PIL, geralmente, é insidioso, com a duração dos sintomas cursando de dois meses até 12 anos antes da procura por ajuda médica; com um período médio de três anos. A doença pode ser diagnosticada incidentalmente, apesar de que a maioria dos pacientes se tornam sintomáticos, com dispneia ao esforço e tosse não produtiva. Sintomas respiratórios estão presentes na maioria dos pacientes no momento do diagnóstico e incluem dispneia e tosse seca. Alguns pacientes também relatam dor pleurítica e sintomas sistêmicos, como febre, perda de peso, fadiga e sudorese noturna.
Diagnóstico:
O lavado broncoalveolar é útil para descartar outros diagnósticos, como infecções concomitantes; entretanto, não tem achados específicos para PIL. A biópsia transbrônquica raramente é conclusiva no diagnóstico da PIL, podendo em raros casos demonstrar a infiltração linfocítica do interstício peribroncovascular típica da PIL; a biópsia cirúrgica é ferramenta diagnóstica amplamente superior e requisitada para confirmação diagnóstica da PIL.
Após estabelecido o diagnóstico histopatológico de PIL, deve-se realizar uma pesquisa aprofundada de doenças autoimunes e imunodeficiências que possam estar relacionadas. Um painel de exames laboratoriais é recomendado, incluindo: anticorpos antinucleares, anticorpo antipeptídeo citrulinado cíclico (anti-CCP), hematimetria, painel metabólico, dosagem de anticorpos contra EBV, HIV e HTLV, dosagem quantitativas de imunoglobulinas, fator reumatoide, eletroforese de proteínas séricas, dosagem de anti-SSA-Ro e anti-SSB-La e estudos de função tireoideana.
Diagnóstico diferencial: A PIL pertence ao grupo de doenças pulmonares caracterizadas pela presença de múltiplos cistos, juntamente com a linfangioliomiomatose (LAM) , histiocitose de células de Langerhans (HCL) , síndrome de Birt-Hogg-Dube (SBHD) , amiloidose e a doença de deposição de cadeias leves (DDCL) .
-
Exames de imagem:
- Radiografia de tórax: Não apresenta achados sensíveis ou específicos para PIL;
- Tomografia computadorizada do tórax: A pneumonia intersticial linfocítica caracteriza-se pela presença de múltiplos cistos com distribuição randômica, isto é, aleatória, em adjacência a vasos sanguíneos. Nódulos pulmonares, usualmente, são achados encontrados em associação, auxiliando no diagnóstico diferencial com outras doenças císticas (figuras acima).
Autor(a) principal: Elazir Mota (Radiologia, especialista em Radiologia Pediátrica).
Lynch JL, Blickman JG, terMeulen DC, et al. Radiographic resolution of lymphocytic interstitial pneumonitis (LIP) in children with human immunodeficiency virus (HIV): not a sign of clinical deterioration. Pediatr Radiol. 2001; 31(4):299-303.
Rajagopala S, Singh N, Gupta K, et al. Pulmonary amyloidosis in Sjogren's syndrome: a case report and systematic review of the literature. Respirology. 2010; 15(5):860-6.
Sirajuddin A, Raparia K, Lewis VA, et al. Primary Pulmonary Lymphoid Lesions: Radiologic and Pathologic Findings. Radiographics. 2016; 36(1):53-70.
Souza JR, Soares A, et al. Terminologia para a descrição de tomografia computadorizada do tórax: Sugestões iniciais para um consenso brasileiro. Radiol Bras. 2002; 35(2):125-128.